















Research themes
Molecular Medical Science Laboratory has a mission to create novel medicines, degraders made from a
small molecular compound turning the undruggable disease causative proteins into druggable targets using
our unique technology free from ubiquichination,CANDDY.
See also our institute's website at http://www.ribs.tus.ac.jp/index.php/course/labforstu/
1. Targets Regulation & Drug Discovery – A novel approach for next-generation molecular target drug based on "CANDDY" technology.
In the personal genomic era, many target proteins involved in diseases has been identified, and has been steadily advanced on basic study for drug discovery. However, the discovery of molecular target drugs targeting those identified protein, has been faced with the problem that it is difficult to "undruggable". Since about 80% of the proteome has been said to be undruggable, development of molecular target drugs, is an important issue for overcoming of the disease. In order to tackle the issue, we attempt to establish a new technology "CANDDY" (=chemical interactome (interactions) and knockdown). CANDDY(protein level in Figure1) technology called "chemical knockdown", which aims to control directly the degradation of target proteins without requirements of any genetic manipulations and DDS unlike siRNA. We focus on the phenomena of proteolysis independent from the ubiquitine system. The goal of our "chemical knockdown" project is to control direct degradation for undruggable targetome proteins.
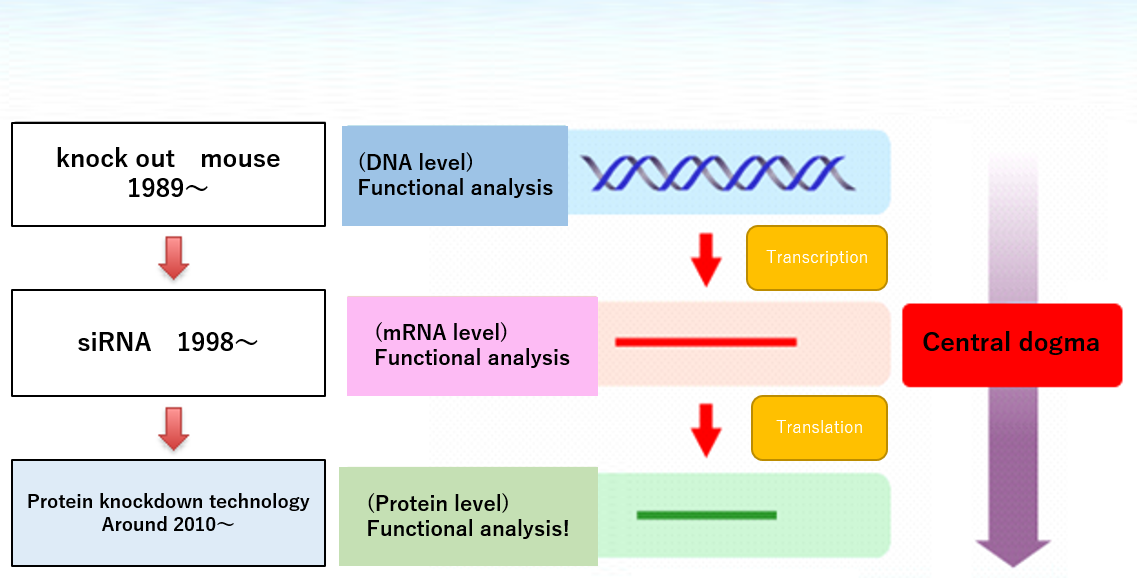
Figure 1. Central dogma in Life Science
2. Targets Determination – Development of "chemical interactome" based on IVV-HiTSeq technology.
What is the interactome?
Although the human genome sequence was successfully determined in the genome project, the functions for more than half of the discovered genes (proteins) were unknown. To determine the protein functions, it is important to find their interaction partners. Proteins always interact with other biomolecules for their biological functions. We call the entirety of these interactions between biomolecules as the 'interactome'. However, to correctly understand dynamically changing interactome networks is not easy. New methods have had to be developed to analyze spatiotemporal networks efficiently. Our challenge is to develop these new technologies for capturing the spatiotemporal networks.
Next-generation sequencing (NGS) has been applied to various kinds of omics studies, resulting in many biological and medical discoveries. We have developed a cell-free display technology combined with NGS that can improve both the coverage and reliability of interactome datasets showed in the figure. The completely cell-free method gives a high-throughput and a large detection space, testing the interactions without using clones. The quantitative information provided by NGS reduces the number of false positives. The method is suitable for the in vitro detection of proteins that interact not only with the bait protein, but also with DNA, RNA and chemical compounds. Thus, it could become a universal approach for exploring the large space of protein sequences and interactome networks. Currently, we attempt to develop "chemical interactome", which is a compound-protein interaction pair screening technique based on the IVV-HiTSeq method.
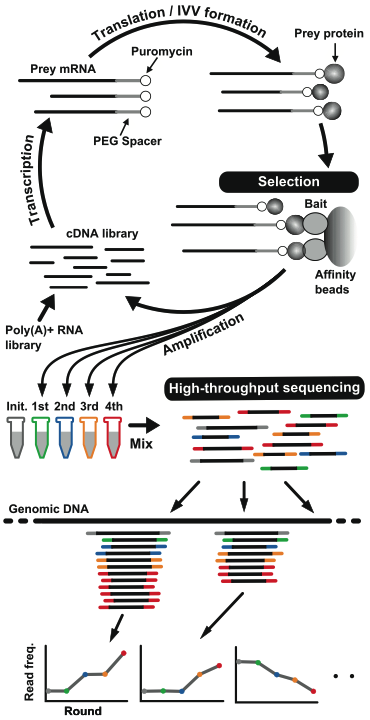
Figure 2. Overview of IVV-HiTSeq
(http://www.natureasia.com/ja-jp/srep/abstracts/40682)
3. Targets Confirmation – Development of a new conditional mouse based on controlling protein knockdown.
In order to clarify the pathogenic mechanism of disease and establish effective therapy, it is urgently necessary for product the animal models which can be analyzed dynamically. To date, it is well known that knockout mouse model and conditional genetically engineered mouse model were developed for analyzing the functions of gene and protein involved in disease. However, these methods have drawbacks, including that the function could not be analyzed due to embryonic lethal, disable to pass through the blood-brain barrier, large side effects, and efficiency is low requiring repetitive exposure to the inducing drugs. In order to tackle the issue, we are creating a novel transgenic mouse model, in which the protein-of-interest (POI) was fused with the destabilizing domain (DD) named DD mouse. Newly synthesized DD-POIs are degraded through the proteosomal pathway, but their decay can be blocked with the ligands trimethoprim (TMP). Therefore, the target protein can be stabilized by TMP in DD mouse. This is different from the conditional mouse, the target protein can be controlled reversibly. Moreover, it is characterized that the amount of target protein can be regulated directly and reversibly, which is different from the genetic regulation indirectly. Thus, embryonic lethality can be avoided, and functional analysis of the target protein will become possible. We aim to clarify the possibility of controlling the target protein quantitatively and reversibly, and to avoid the embryonic lethality of the DD-POI homozygous controlled by TMP administration in this novel mouse model.
About Our Research
Puromycin technology developed in Japan
Around 1995, some researchers reported that ribonucleic acids (RNA) could acquire functions by directed evolution in a test tube ("in vitro evolution"). The next target of in vitro evolution was to make proteins evolve faster into preferred forms in the test tube. As a result, we succeeded in developing the in vitro virus (IVV) method (Figure 1A) as a research tool for analyzing the in vitro evolution of proteins. In addition, we developed a method of labeling the C-terminus of a protein using puromycin (Figure 1B). This method was fortuitously discovered while developing the IVV method. We named these two protein analysis methods "puromycin technology".
Figure 1. Puromycin Technology
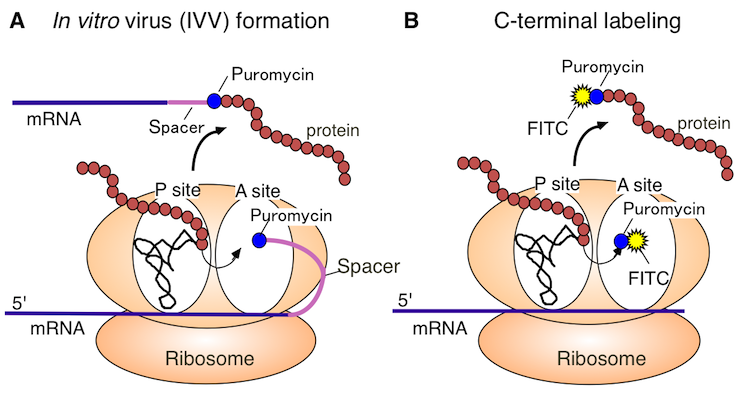
A. The figure is a schematic representation of the in vitro virus (IVV) formation. In vitro virus (IVV) is a fusion molecule of mRNA (genotype) and encoded protein (phenotype) covalently linked via puromycin and a polyethylene glycol spacer. The reaction of the IVV formation is catalyzed by the ribosome-centered translational machinery in a cell-free system.
B. The figure is a schematic representation of the protein C-terminal labeling procedure. Under moderately low puromycin concentration conditions, puromycin derivatives (e.g., fluorescent puromycin conjugate) bind to the C-terminus of the full-length protein. This reaction is catalyzed by the ribosome-centered translational machinery in a cell-free system.
Functional analysis in the post-genomic era: The world's first analysis tool for region-level protein interactions
In 2000, when the research on puromycin technology was still in its infancy (Miyamoto-Sato et al., NAR, 2000), a historical event in life science took place. In June of that year, Dr. Francis Collins, a director of international human genome project (HGP), formally declared that mapping of 90% of the regions of the human genome was completed. Scientists worldwide were excited by this announcement, which heralded the opening of the 'post-genomic' era for unraveling the code written in the genomic sequences. In response to this, the Japanese government started some national projects related to the human genome.
At the same time, we started applying the puromycin technology to the functional analysis of proteins in the post-genomic era. In studies dealing with a large amount of data like genomic sequences, not only experimental analysis tools, but also interdisciplinary approaches, including bioinformatics, were needed. In 2005, we succeeded in developing a high-throughput system for protein interaction analysis through collaborative research with the IT company (Miyamoto-Sato et al., Genome Res., 2005).
Yeast two-hybrid (Y2H) system and TAP (Tandem Affinity Purification)-MS (mass spectrometry) methods are well-known PPI analysis methods. However, these methods suffer from certain problems, such as cell toxicity and the library size derived from cell manipulations in their experimental procedures. Our methods can overcome these problems.
In the genome network project (promoted as a national project in Japan), we developed an automated system that could conduct large-scale experiments using the IVV method (Figure 2-left). Using this system, we succeeded in the world's first region-/domain-level interaction analysis of human transcription factors (Figure 2-right; Miyamoto-Sato et al., PLoS One, 2010). The data is freely available on the website (Genome Network Platform) of National Institute of Genetics.
Figure 2. The analysis of transcription factor networks using the IVV method
